
RESEARCH
Lipid droplet turnover at the lysosome inhibits growth of hepatocellular carcinoma in a BNIP3-dependent manner
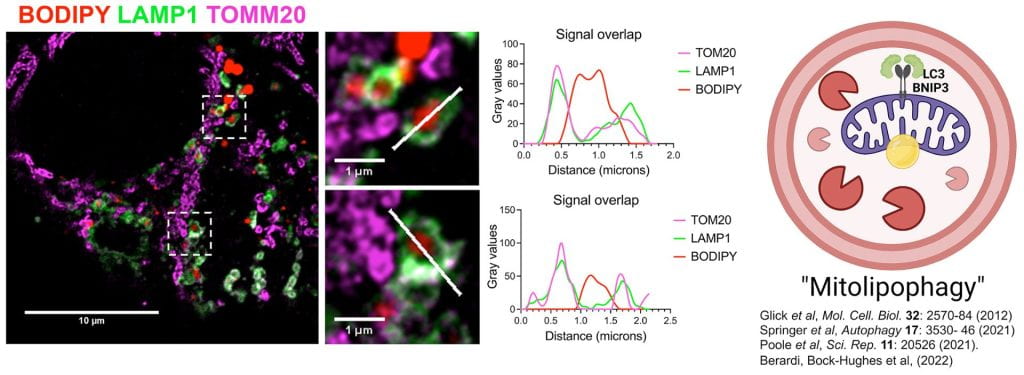
Hepatic steatosis is a major etiological factor in hepatocellular carcinoma (HCC, but factors causing lipid accumulation leading to HCC are not understood. We identify BNIP3 (a mitochondrial cargo receptor) as an HCC suppressor that mitigates against lipid accumulation to attenuate tumor cell growth. Targeted deletion of BNIP3 decreased tumor latency and increased tumor burden in a mouse model of HCC. This was associated with increased lipid in BNIP3-/- HCC at early stages of disease while lipid did not accumulate until later in tumorigenesis in wild-type mice, as BNIP3 expression was attenuated. Low BNIP3 expression in human HCC similarly correlated with increased lipid content and worse prognosis than HCC expressing high BNIP3. BNIP3 suppressed HCC cell growth by promoting lipid droplet turnover at the lysosome in a manner dependent on BNIP3 binding LC3. We have termed this process “mitolipophagy” since it involves the coordinated autophagic degradation of lipid droplets with mitochondria.
Highlights
- Loss of BNIP3 promotes hepatic steatosis in the pericentral zone of liver.
- Hepatic steatosis associated with aging correlates with decreased BNIP3 expression.
- BNIP3 promotes lipid droplet turnover at the autolysosome.
- Loss of BNIP3 causes premature hepatic steatosis and promotes hepatocellular carcinoma.
ULK1 promotes mitophagy via phosphorylation and stabilization of BNIP3
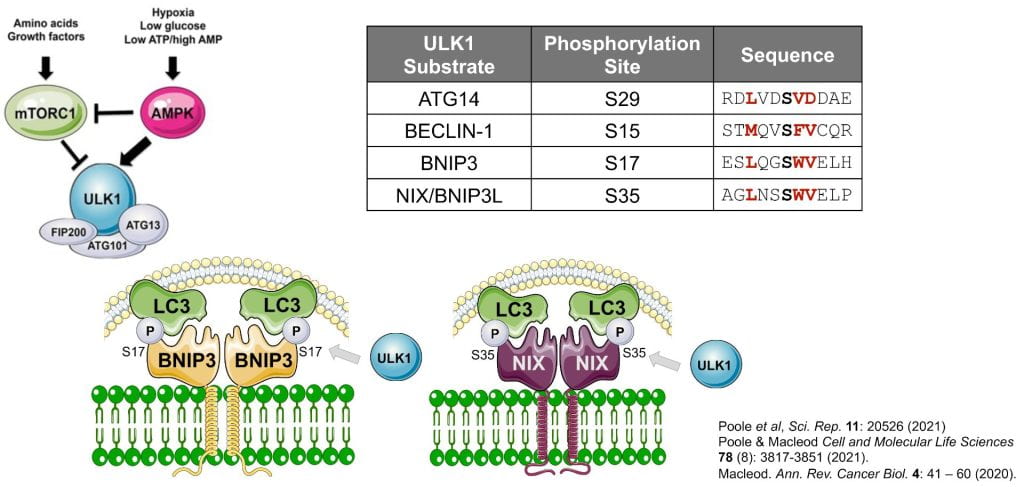
UNC51-like kinase-1 (ULK1) is the catalytic component of the autophagy pre-initiation complex that stimulates autophagy via phosphorylation of ATG14, BECLN1 and other autophagy proteins. ULK1 has also been shown to specifically promote mitophagy but the mechanistic basis of how has remained unclear. Here we show that ULK1 phosphorylates the BNIP3 mitochondrial cargo receptor on a critical serine residue (S17) adjacent to its amino terminal LIR motif. ULK1 similarly phosphorylates BNIP3L on S35. Phosphorylation of BNIP3 on S17 by ULK1 promotes interaction with LC3 and mitophagy. ULK1 interaction also promotes BNIP3 protein stability by limiting its turnover at the proteasome. The ability of ULK1 to regulate BNIP3 protein stability depends on an intact “BH3” domain, and deletion of its “BH3” domain reduces BNIP3 turnover and increases BNIP3 protein levels independent of ULK1. In summary, ULK1 promotes mitophagy by both stabilization of BNIP3 protein and via phosphorylation of S17 to stimulate interaction with LC3.
Highlights
- BNIP3 and NIX are phosphorylated on S17 and S35 respectively by ULK1.
- Phosphorylation of BNIP3 and NIX by ULK1 promotes their interaction with processed LC3 and mitophagy.
- Interaction of BNIP3 with ULK1 also increases BNIP3 protein levels by inhibiting proteasomal degradation of BNIP3 protein, further increasing rates of mitophagy.
BNIP3-dependent mitophagy promotes cytosolic localization of LC3B and metabolic homeostasis in the liver
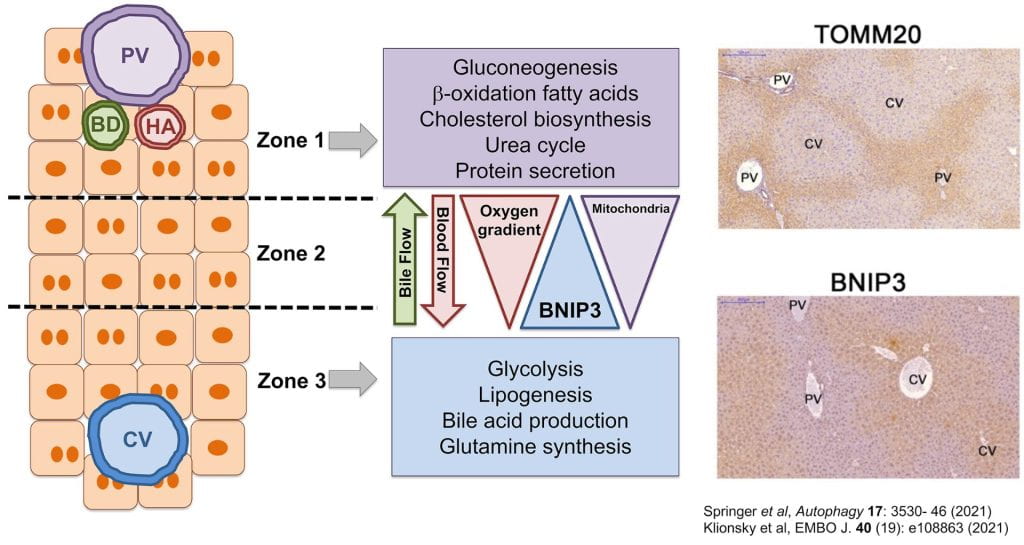
Mitophagy formed the basis of the original description of autophagy by Christian de Duve when he demonstrated that GCG (glucagon) induced macroautophagic/autophagic turnover of mitochondria in the liver. However, the molecular basis of liver-specific activation of mitophagy by GCG, or its significance for metabolic stress responses in the liver, is not understood. Here we show that BNIP3 is required for GCG-induced mitophagy in the liver through interaction with processed LC3B—an interaction that is also necessary to localize LC3B out of the nucleus to cytosolic mitophagosomes in response to nutrient deprivation. Loss of BNIP3-dependent mitophagy caused excess mitochondria to accumulate in the liver, disrupting metabolic zonation within the liver parenchyma, with expansion of zone 1 metabolism at the expense of zone 3 metabolism. These results identify BNIP3 as a regulator of metabolic homeostasis in the liver through its effect on mitophagy and mitochondrial mass distribution.
Highlights
- BNIP3 is required for glucagon-induced mitophagy in liver.
- BNIP3 is expressed in a zonal pattern across the liver lobule.
- Loss of BNIP3 disrupts zonal metabolism in the liver.
Autophagy in tumor progression to metastasis
Autophagy is a conserved catabolic process that plays a housekeeping role in eliminating protein aggregates and organelles and is activated during nutrient deprivation to generate metabolites and energy. Autophagy plays a significant role in tumorigenesis, although opposing context-dependent functions of autophagy in cancer have complicated efforts to target autophagy for therapeutic purposes. We demonstrate that autophagy inhibition reduces tumor cell migration and invasion in vitro and attenuates metastasis in vivo. Numerous abnormally large focal adhesions (FAs) accumulate in autophagy-deficient tumor cells, reflecting a role for autophagy in FA disassembly through targeted degradation of paxillin. We demonstrate that paxillin interacts with processed LC3 through a conserved LIR motif in the amino terminal end of paxillin and that this interaction is regulated by oncogenic SRC activity. Together, these data establish a function for autophagy in FA turnover, tumor cell motility, and metastasis.

Highlights
- Autophagy is required for the migration and invasion of metastatic tumor cells.
- Autophagy promotes the degradation of paxillin and focal adhesion turnover.
- Paxillin interacts with LC3B through a conserved LIR in a Src-regulated manner.
- Autophagy is required for Src-regulated tumor cell motility.
- These studies validate targeting autophagy to inhibit metastatic disease.
BNIP3 and mitophagy control in breast cancer
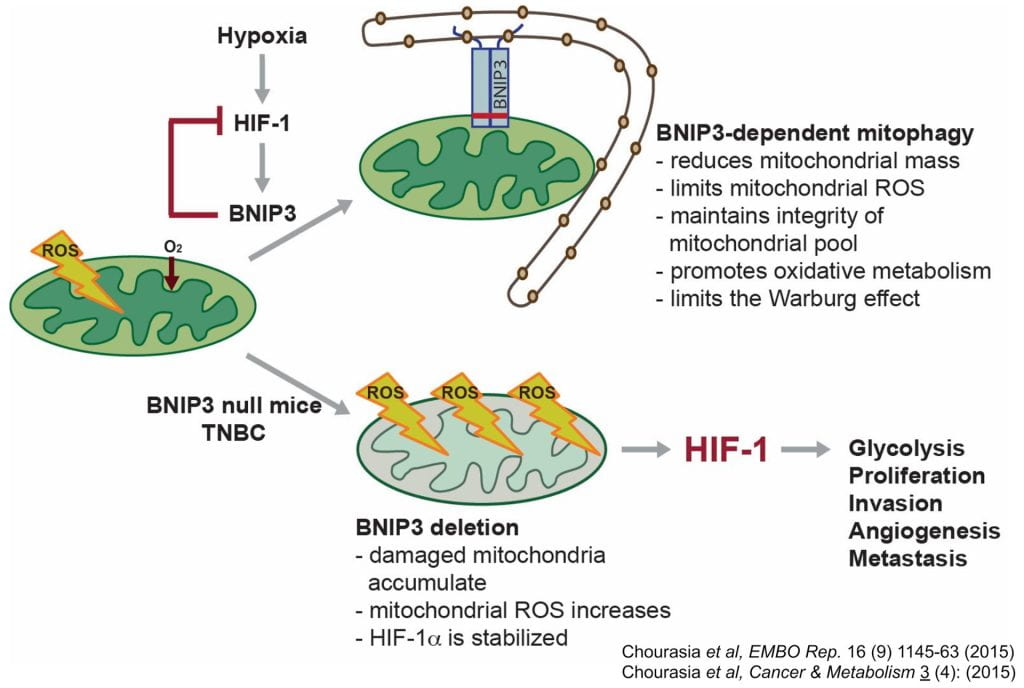
BNIP3 is a hypoxia-inducible protein that targets mitochondria for autophagosomal degradation. We have identified a novel tumor suppressor role for BNIP3 in a clinically relevant mouse model of mammary tumorigenesis. We have demonstrated a functional link between the activity of BNIP3 in retarding primary mammary tumor growth and progression to preventing the accumulation of dysfunctional mitochondria and resultant excess ROS production. In the absence of BNIP3, mammary tumor cells are unable to reduce mitochondrial mass effectively and increased mitochondrial ROS caused elevated expression of Hif-1α and Hif target genes, including those involved in glycolysis and angiogenesis–two processes that are also markedly increased in BNIP3 null tumors. Glycolysis inhibition attenuated growth of BNIP3 null tumor cells, revealing increased dependence on autophagy for survival. Our data also demonstrates that BNIP3 deletion can be used as a prognostic marker of tumor progression to metastasis in human triple negative breast cancer (TNBC). Our work reveals how mitochondrial dysfunction due to defects in mitophagy can promote the Warburg effect and tumor progression, and also suggests better approaches to stratifying TNBC for treatment.
Highlights
- Loss of BNIP3 promotes tumor cell growth and progression to metastasis in the MMTV-PyMT mammary tumor model;
- Elevated ROS production by dysfunctional mitochondria in BNIP3 null tumors results in increased Hif-1α levels and increased tumor progression to invasiveness;
- This defines a novel negative feedback loop between BNIP3 and Hif-1a that limits the oncogenic activity of Hif-1 in glycolysis and angiogenesis;
- Defective mitochondria and increased aerobic glycolysis arising from loss of BNIP3 induces greater dependence on autophagy for survival;
- BNIP3 is focally deleted in triple negative breast cancer and together with high HIF-1a levels strongly predicts progression to metastasis in TNBC patients.